文章目录
-
- 写在前面
- 读取SAM文件
- 读取FASTA文件
参考官网pysam API:https://pysam.readthedocs.io/en/latest/api.html
属性常用的一些属性,读取SAM文件、FASTA文件。
读取SAM文件
读取sam文件时,熟悉一些常用的属性。
导入pysam模块
import pysam
使用pysam读取sam/bam文件,并打印一条read信息:
mysam = '/mydir/xxx.bam'
sam_rd = pysam.AlignmentFile(mysam, 'rb') # bam文件,指定使用二进制读取
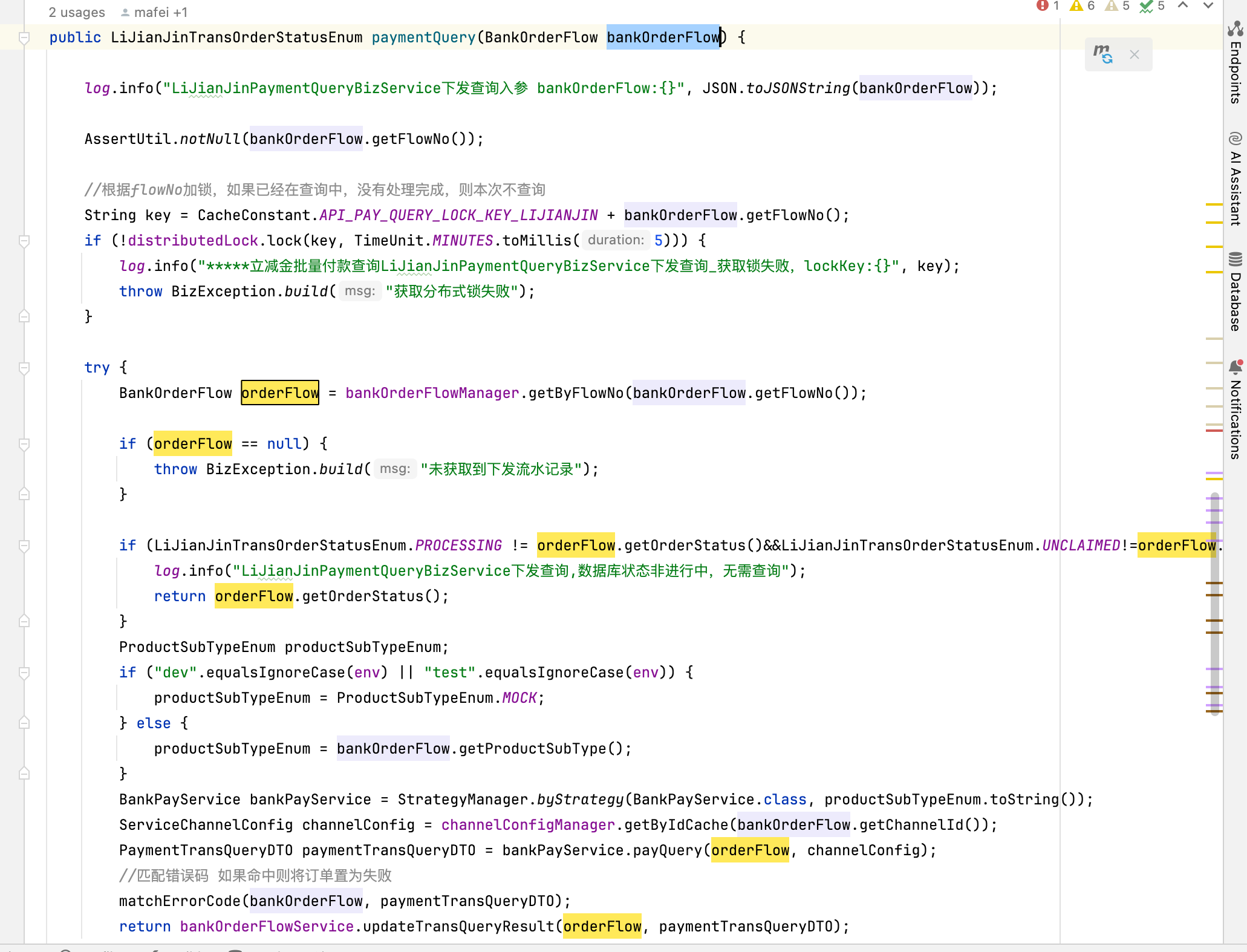
for rec in sam_rd:
print(rec)
break
sam_rd.close()
# 或用with, 就不需要写close()
with pysam.AlignmentFile(mysam, 'rb') as sam_rd:
for rec in sam_rd:
print(rec)
break
使用pysam写入一条read信息到sam/bam文件:
outsam = '/mydir/out.bam' sam_wrt = pysam.AlignmentFile(outsam, 'wb', template=sam_rd) # wb是使用二进制写入;template模板header部分使用sam_rd中的头部header sam_wrt.write(rec) # rec是sam_rd读取的一条record sam_wrt.close() # 或用with, 就不需要写close() with pysam.AlignmentFile(outsam, 'wb', template=sam_rd) as sam_wrt: sam_wrt.write(rec)
- 获取sam各列信息:
# 示例: print(rec.query_name) # 获取第一列,readID print(rec.flag) # 获取第二列,FALG值 print(rec.reference_name) # 获取第三列,reference名称
列 说明 使用pysam属性 1 readID名 rec.query_name 2 FALG值 rec.flag 3 参考序列名称 rec.reference_name 4 比对位置 rec.pos 5 比对质量值 rec.mapping_quality 6 CIGAR值 rec.cigar 7 配对read的参考序列名称 rec.next_reference_name 【对PE序列有效】 8 配对read的比对位置 rec.next_reference_start 【对PE序列有效】 9 序列模板长度 rec.template_length 10 序列 rec.query_sequence 11 比对质量值 rec.qual 之后 候选字段标签tag rec.tags(返回tuple类型)
# 更多相似属性: print(rec.reference_id) # 得到的是从0开始的数值(对应是按构建fasta文件的名称顺序) print(rec.next_reference_id) # 配对read的参考序列id,类似rec.reference_id print(rec.positions) # 获取比对上的碱基对应的所有位置,比如: [10,11,12,13,14] print(rec.cigarstring) # 和rec.cigar同,建议用rec.cigarstring print(rec.cigartuples) # 获取tuple类型,比如:8S10M 对应 [(4, 8), (0, 10)] print(rec.query_alignment_start) # 比对的相对起始位置(比如从第1bp开始比对,则为0;比如前5bp是softclip,则开始位置是从第6bp碱基开始,则这里输出位置为5; print(rec.query_alignment_end) # 比对的相对终止位置 print(rec.query_alignment_sequence) # 比对上的序列,这里不输出softclip对应的碱基 print(rec.query_alignment_qualities) # 比对上的碱基质量值数值格式(Phred33) print(rec.query_alignment_length) # 比对上的序列长度 print(rec.seq) # 同rec.query_sequence,建议用rec.query_sequence print(rec.query) # 输出的同rec.seq? print(rec.qqual) # 输出的同rec.qual? print(rec.query_qualities) # 序列的碱基质量数值格式(Phred33) print(rec.query_length) # 序列长度
-
如何将读取的信息转换成字符?
https://pysam.readthedocs.io/en/latest/api.html?highlight=tostring#pysam.AlignedSegment.to_string
print(rec) # 直接输出的bam信息,比如位置信息是0-based print(rec.tostring(sam_rd)) # 相当于直接输出为sam格式的信息(位置信息是1-based)
注:rec.tostring(sam_rd)的写法是否有问题?
-
判定该记录序列信息是read1或read2,是否是反向比对?是否成对?
if rec.is_read1: print('rec is read1') if rec.is_read2: print('rec is read2') if rec.is_reverse: print('rec is reverse alignment') if rec.is_paired: print('rec is paired') -
判定该记录序列信息是否未比对上?是否是二次比对?是否是补充比对?
if rec.is_unmapped: print('rec is unmapped') if rec.is_secondary: print('rec is secondary alignment') if rec.is_supplementary: print('rec is supplementary alignment') -
配对序列是否反向比对?配对序列是否未比对上?
if rec.mate_is_reverse: print('mate read is reverse alignment') if rec.mate_is_unmapped: print('mate read is unmapped')
读取FASTA文件
使用pysam处理fasta文件,示例fasta文件:
$ cat /mydir/xxx.fasta >chr1 ATCGAG >chr2 GCATCGAA
使用pysam读取fasta文件:
fa = '/mydir/xxx.fasta'
read_fa = pysam.FastaFile(fa)
print(read_fa.references) # fasta参考序列名称list
print(read_fa.nreferences) # fasta参考序列有几个
print(read_fa.lengths) # fasta参考序列长度
print(read_fa.fetch('chr1')) # 获取名为chr1的参考序列所有碱基
print(read_fa.fetch('chr1', 2, 5)) # 获取chr1参考序列的第3个到第5个碱基
read_fa.close()
输出内容:
['chr1', 'chr2'] 2 [6, 8] ATCGAG CGA
注:如果没有建立索引,使用pysam会自动建立索引文件(后缀.fai) ,或使用samtools faidx /mydir/xxx.fasta 建立。
未完待续。。。